
Beschreibung
|
|
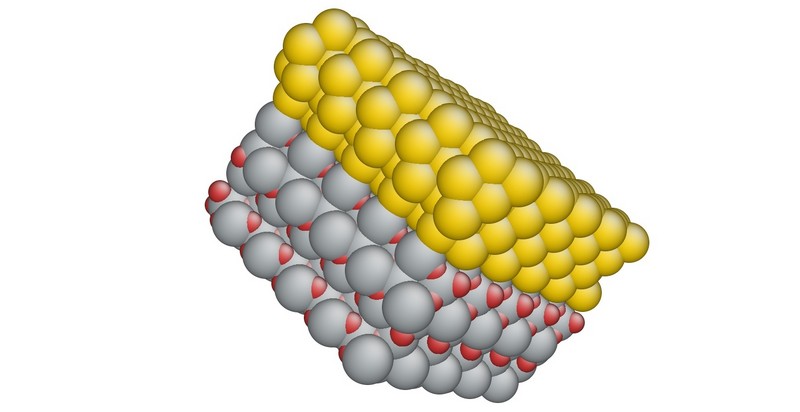
Viele materialwissenschaftliche Probleme erfordern die Simulation zusammengesetzter Systeme aus Oxiden, Metallen und Halbleitern (siehe Abbildung 1). Im Teilprojekt B.1 sollen Wechselwirkungspotenziale für Oxide entwickelt und in unseren Code IMD implementiert werden. Damit werden wir Oxid-Metall-Grenzflächen, insbesondere die Mechanismen des Materialversagens unter mechanischer und thermischer Belastung, untersuchen.
Gegenstand des Teilprojekts ist die Simulation mehrkomponentiger nanoskaliger Systeme. In Halbleiterbauteilen etwa werden Isolatorschichten aus Oxiden im Kontakt mit metallischen Leiterbahnen benötigt. Das Verhalten solch zusammengesetzter Systeme unter mechanischer und thermischer Belastung ist von höchster Anwendungsrelevanz. Die gängigen Molekulardynamik-Codes unterstützen Wechselwirkungspotenziale für oxidische Materialien in der Regel nicht gleichzeitig mit Wechselwirkungen für Metalle und Halbleiter.
Deshalb soll unser Molekulardynamik-Code IMD in die Lage versetzt werden, zusätzlich zu Metallen und Halbleitern auch Wechselwirkungen für oxidische Materialien zu behandeln. Zu diesen tragen auch weit reichende Coulomb- und Dipolkräfte bei. Das zu implementierende Potenzialmodell soll einerseits für quantitativ korrekte Simulationen hinreichend präzise und gut übertragbar sein. Andererseits sollen die Kräfte aber auch so effizient und gut skalierbar zu berechnen sein, dass man damit Systeme von mehreren Millionen Atomen routinemäßig simulieren kann. Erforderliche Kraftfelder werden mit unserem Potenzial-Modellierungs-Code potfit generiert, welcher um die neuen Verfahren erweitert werden kann.
Unerlässlich ist auch die korrekte Behandlung der Ladungsverteilung an einer Grenzschicht. Die Valenzen in einem Oxid bauen sich zur Grenzschicht mit dem Metall hin ab. Es gibt dazu verschiedene Verfahren, wie z.B. das unten beschriebene von Streitz und Mintmire.
Ziel ist es, Simulationen oxidischer Materialien sowie kombinierter Metall-Metalloxid-Systeme in materialwissenschaftlichen Dimensionen zu ermöglichen, wie dies heute schon mit reinen Metallen oder kovalent gebundenen Systemen üblich ist.
Stand der Forschung
Für Metalle und kovalente Materialien sind bereits in gängigen Molekulardynamik-Codes (insbesondere in IMD) realisiert:
-
Systemgrößen > 10^7 Atome
-
Simulationszeiten bis Nanosekunden durch gute Skalierbarkeit in der CPU-Zahl
-
genaue und effiziente Potenzialmodelle
Die Herausforderung besteht darin, ähnliche Zieldaten auch für Metall-Oxide zu erreichen. Zur effizienten Behandlung weit reichender elektrostatischer Wechselwirkungen gibt es
die sogenannte Wolf-Summation [1], eine direkte Summierung mit linearen Skalierungseigenschaften. Ein Wechselwirkungsmodell zur Berücksichtigung der Polarisierbarkeit einzelner Ionensorten wurde von Tangney und Scandolo [2] entwickelt.
Um ein Metall in Kontakt mit seinem Oxid in der Simulation darzustellen, kann das Streitz-Mintmire-Verfahren [3] eingesetzt werden. Die Valenzen werden dabei durch Minimierung der elektrostatischen Energie bestimmt. Damit können dann gerade elektrostatische Grenzflächeneffekte korrekt beschrieben werden.
Literatur
[1] D. Wolf, P. Keblinski, S. R. Phillpot, und J. Eggebrecht, J. Chem. Phys. 110, 8254 (1999).
[2] P. Tangney und S. Scandolo, J. Chem. Phys. 117, 8898 (2002).
[3] F. H. Streitz und J. W. Mintmire, Phys. Rev. B 50(16), 11996 (1994).