
Beschreibung
|
|
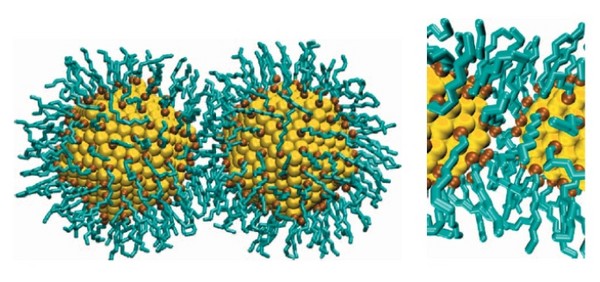
Nanokristalle aus Gold oder Halbleitermaterialen können zu einer Reihe unterschiedlicher zwei- und dreidimensionaler Superstrukturen selbstassemblieren. Die sich ausbildenden Kristallstrukturen können sehr unterschiedlich sein, und hängen zum Beispiel von den Größenverhältnis der Primärkristalle ab, aber auch von den Lösungsmitteln und den die Lösung stabilisierenden tensidischen Liganden.
Da die Erzeugung einer speziell geeigneten Kristallstruktur wichtig ist für viele Anwendungen, ist das Ziel des Projektes die Entwicklung einer geeigneten Simulationsmethodik, um das Phasendiagramm der Kristallphasen einer monodispersen oder bidispersen Lösung aus Gold-Nanokristallen als Funktion
der Größenverhältnisse der Nanokristalle, des Lösungsmittels, der Temperatur, und der der Ligandenlänge zu bestimmen. Dazu sollen zuerst explizite atomistische Molekulardynamiksimulationen eines Systems bestehend aus zwei oder drei Nanokristallen im expliziten Lösungsmittel durchgeführt werden. Aus diesen Simulationen werden effektive Paar- und Dreikörperwechselwirkungspotenziale für eine anschließende lösungsmittelfreie Simulation bestimmt.
Wegen der flexiblen Liganden spielen Dreikörperpotenziale für die Wechselwirkungen zwischen Nanokristallen eine wichtige Rolle. Mittels dieser effektiven Potenziale sollen dann Vielteilchensysteme in einer vergröberten Simulation untersucht werden, mit dem Ziel, das Phasendiagramm auszurechnen. Dazu wird die freie Energie der Kristallstrukturen mittels berechnet. Die erhaltenen Ergebnisse sollen mit experimentellen Daten aus der Literatur verglichen werden, damit man die Genauigkeit der entwickelten Methode abschätzen kann. Die Ergebnisse werden auch Einblicke erlauben, unter welchen Bedingungen Dreikörperpotenziale eine große Rolle spielen.