
Description
|
|
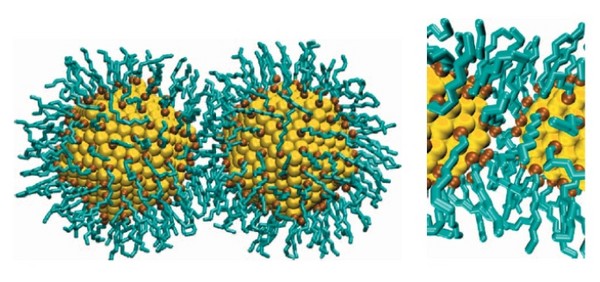
Gold nanocrystals coated with ligands can self-assemble in a variety of two and three dimensional superstructures. The obtained crystal structures depend not only on external conditions like temperature or the type of the solvent, but also the characteristics of the capping ligands play an important role. Since for many applications the creation of some certain crystal structure is of crucial importance, the aim of this project is to develop a simulation methodology to obtain a phase diagram of crystal structures for mono and bi-disperse gold nanocrystals depending on the size of the gold cores, length of ligands, temperature and a type of the solvent.
This is planned to be achieved in several steps:
- Carry out explicit atomistic molecular dynamics simulations of two nanocrystals to obtain the effective interaction between the nanocrystals depending on the size of the gold cores, length of ligands, temperature and a type of the solvent.
- Add a third nanocrystal to obtain also three-body potentials. Due to the flexible ligands and crowding effects, the three-body interaction should play an important role.
- Using the obtained coarse-grained two and three body potentials, calculate the free energy of different crystal structures and acquire the phase diagram of the system.
Our results will be compared with experimental results that are available for certain parameter combinations, so that one can judge the reliability and the precision of the methodology. This will also allow to estimate under which conditions the three-body interactions have to be taken into account. Using our method, we can then use computer simulations to systematically extend the phase diagram into regions where no experiments are yet available.