
Ergebnisse
In der ersten Förderphase wurden Molekulardynamiksimulationen zur Untersuchung des Bruchs der spröd/duktil-Modellsysteme Ni/Ni3Al und Ni/NiAl durchgeführt. Es wurden Zugversuche simuliert, mit denen die Spannungs-Dehnungs-Kurven in Abhängigkeit von Grenzflächenorientierung, Misfitversetzungen und Temperatur bestimmt wurden.
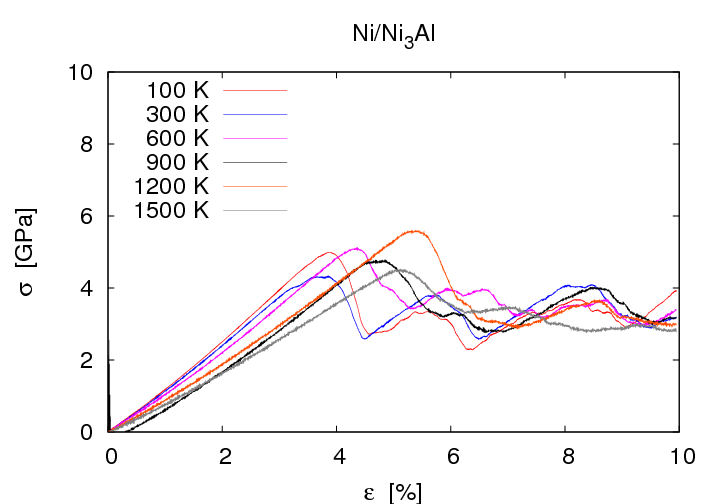
Das System Ni/Ni3Al zeigt durchgehend duktiles Materialversagen, es tritt auch bei hohen Dehnungen kein Riss auf. Erhöht man die Dehnung über die Zugfestigkeit hinaus, so bilden sich die für fcc-Kristalle typischen intrinsischen Stapelfehler, die von Shockley-Partialversetzungen umrandet sind. Während in den Einkristallen sowie im System ohne Misfitversetzungen homogene Versetzungsbildung auftritt, zeigt sich beim System mit Misfitversetzungen der deutliche Einfluss dieser Defekte auf die Versetzungsentstehung. Die Stapelfehler gehen grundsätzlich von den Misfitversetzungen aus und breiten sich zunächst in Ni aus. Bei weiterer Erhöhung der Dehnung breiten sie sich auch in Ni3Al aus, auch hier beginnend an den Misfitversetzungen. Die Zugfestigkeit ist im System mit Misfitversetungen deutlich geringer, sie beträgt etwa 5 GPa, der Wert für Ni3Al ohne Misfitversetzungen liegt bei 13 GPa.
|
|
|

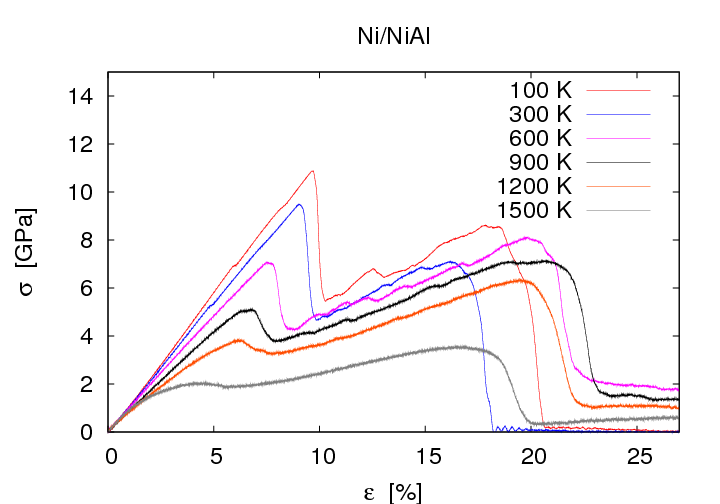
Im System Ni/NiAl zeigen die Spannungs-Dehnungs-Kurven eine deutliche Abhängigkeit von der Orientierung der Grenzfläche. Im Vergleich zur (001)-Grenzflächenorientierung liegt die Zugfestigkeit bei (011)-Grenzflächenorientierung um etwa 4 GPa niedriger, jene bei (111)-Grenzflächenorientierung um etwa 2 GPa höher. Auch bezüglich des Verhaltens bei weiterer Erhöhung der Dehnung gibt es Unterschiede. Bei (001)-Grenzflächenorientierung erfolgt grundsätzlich ein Riss entlang der Grenzfläche, bei (011)- oder (111)-Grenzflächenorientierung gibt es keine Risse, stattdessen nimmt die Defektdichte im duktilen Material weiter zu.
|
|
|
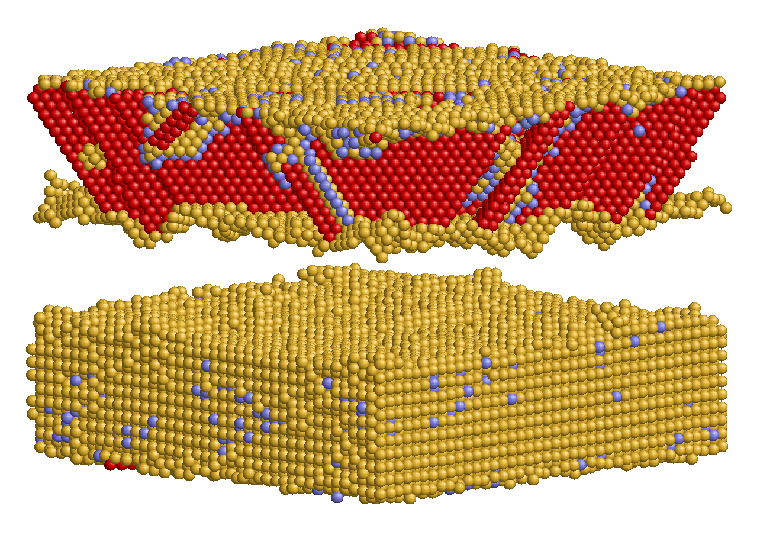
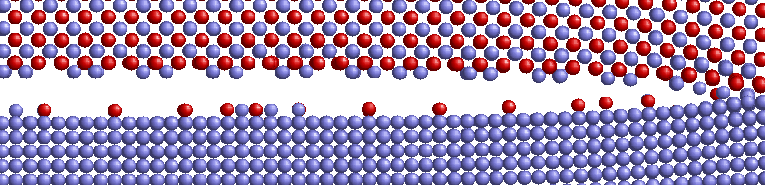
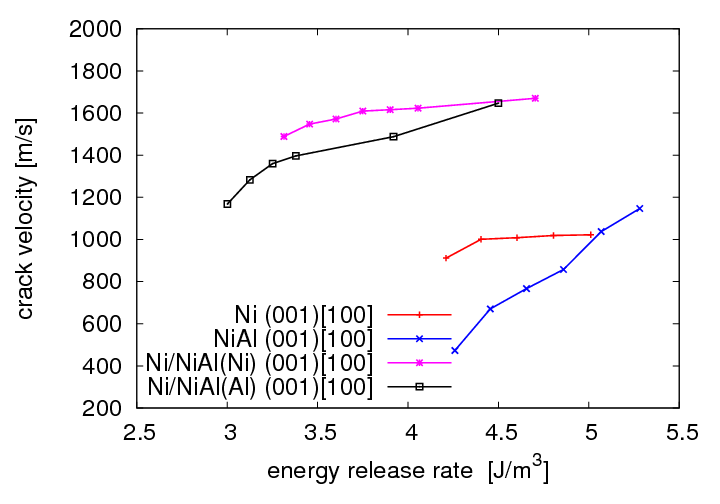
Die Beschaffenheit der Rissfläche sowie die Geschwindigkeiten der Rissausbreitung in der (001)-Grenzfläche wurden analysiert, indem ein Verschiebungsfeld in die Struktur eingebracht wurde und mit Fixed-Displacement-Randbedingungen simuliert wurde.
Zum Vergleich wurden analoge Simulationen in Ni und NiAl Einkristallen durchgeführt. Bezüglich der Beschaffenheit der Rissfläche gibt es einen deutlichen Unterschied zwischen den Systemen mit Al oder Ni an der Grenzfläche. Beim System mit Al an der Grenzfläche kommt es zu einer Auftrennung beider Grenzschichten, es haften sowohl Al-Atome am Ni als auch Ni-Atome am NiAl. Endet NiAl hingegen mit einer Ni-Schicht, so verläuft der Riss glatt in der eingebauten Ebene. Im Gegensatz zum Verhalten der Einkristalle ergibt sich im System mit Ni/NiAl-Grenzfläche ein breiterer Bereich von Energiefreisetzungsraten, bei denen Rissausbreitung stattfindet. Erst ab über 2,0 G0 kommt es anstatt Rissausbreitung zur Erzeugung von Defekten im Ni. Für die Geschwindigkeiten der Risse in der Grenzfläche wurden, sowohl bei [100] als auch bei [110] Ausbreitungsrichtung, im Vergleich zu den Rissen in den Einkristallen deutlich höhere Werte erhalten. Die Rissgeschwindigkeiten in der Grenzfläche liegen im Bereich von 1200-2000 m/s und liegen somit knapp unterhalb der Geschwindigkeiten der Rayleighwellen in den einzelnen Materialien.
|
|
|
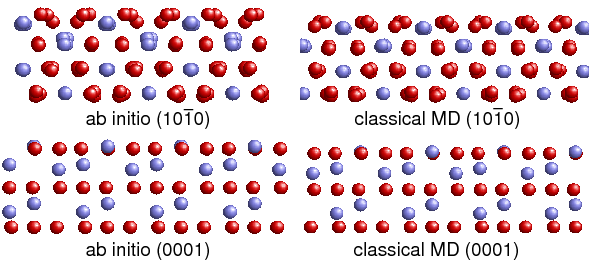
In der zweiten Förderphase wurden in Kooperation mit dem Teilprojekt B.1 interatomare Wechselwirkungspotentiale für α-Al2O3 und Al(111)/Al2O3(0001)-Grenzflächen entwickelt. Es wurden Potentiale erhalten, die Gitterkonstanten, Kohäsionsenergien sowie Oberflächenenergien von α-Al2O3 gut wiedergeben. Auch bezüglich Spannungen gedehnter Konfigurationen sowie der Struktur relaxierter Al2O3-Oberflächen ergeben die neuen Potentiale deutlich realistischere Ergebnisse als bisher für die Molekulardynamiksimulation von Al2O3 verwendete Potentiale. Die mit den Grenzflächenpotentialen berechneten Separationsarbeiten von Al(111)/Al2O3(0001)- Grenzflächenstrukturen liegen nah an den Werten aus der Ab-initio-Berechnung.
|
|
Die neu entwickelten Potentiale wurden in Molekulardynamiksimulationen zur Untersuchung der Rissausbreitung in α-Al2O3 eingesetzt. Es wurden Risse in verschiedenen Orientierungen eingebracht und untersucht, in welchen kristallographischen Ebenen die Rissausbreitung stattfindet. Hierbei ergab sich, dass keine Rissausbreitung in den dichtest gepackten {0001}-Ebenen stattfindet. Stattdessen biegt der Riss in eine {101̅2}-Spaltebene ab. In den {̅2010}-Ebenen hingegen wurde Rissausbreitung beobachtet. Bei einem Startriss in einer { 101̅0}-Ebene verläuft der Rissfortschritt teilweise in dieser Ebene und teilweise in einer {101̅2}-Spaltebene. Diese Ergebnisse stimmen gut mit Resultaten aus Experimenten überein.
Die Ergebnisse der Rissausbreitungssimulationen wurden in Kooperation mit den Teilprojekten B.1 und D.3 visualisiert. Die Resultate für die (̅2010)[0001]- und die (0001)[0̅110]-Orientierungen sind in Abb.8 dargestellt. Hierbei wurden nur die Sauerstoffatome dargestellt und zwar als Pfeil dessen Länge ein Maß für das Dipolmoment des entsprechenden Atoms ist. Mit den Pfeilfarben werden die unterschiedlichen Orientierungen der Dipolmomente verdeutlicht. Zudem ist anhand der Hintergrundfarbe ersichtlich, inwieweit die Dipolmomente in der Umgebung bevorzugte Orientierungen aufweisen. Dadurch sind Regionen mit ausgerichteten Dipolmomenten und solche mit zufällig orientierten Dipolmomenten in der Visualisierung gut erkennbar. In den Ergebnissen der Rissausbreitungssimulationen (Abb.8) fällt auf, dass die Dipolmomente im gedehnten Bereich vor der Rissspitze ausgerichtet sind. Hingegen ergab die Simulation von Al2O3 unter homogener Dehnung keine ausgerichteten Dipole. Die bei den Rissen beobachtete Ausrichtung ist darauf zurückzuführen, dass ein Dehnungsgradient vorliegt. Die als Flexoelektrizität bekannte elektrostatische Polarisation durch Dehnungsgradienten wurde im Folgenden von Teilprojekt B.1 an verschiedenen Oxiden näher untersucht. Hierzu wurden von Teilprojekt B.2 auch Rissausbreitungssimulationen in Magnesiumoxid durchgeführt.
|
|

Des Weiteren wurden mittels Ab-initio-Berechnungen Spannungs-Separations-Gesetze bestimmt, die in der Finite-Elemente-Simulation mit dem Kohäsivzonenmodell eingesetzt werden können. Im verwendeten Verfahren wird die Separation manuell in die Strukturen eingebracht. Durch Relaxation der oberflächennahen Atome wird die potentielle Energie (E) des Systems in Ahängigkeit der Separation (s) berechnet. Daraus wird die Spannung-Separations-Beziehung gemäß σz= 1/A Δ E/Δ s berechnet. Die erhaltenen Spannungs-Separations-Beziehungen für die technologisch relevanten Grenzflächensysteme Metall(111)/Al2O3(0001) (Metall = Al, Cu, Ag, Nb) bei Separation senkrecht zur Grenzfläche sind in Abb.9 dargestellt. Die Maximalspannungen liegen für die kfz-Metalle im Bereich von zwei bis acht GPa, während sie für Niob etwa 27 GPa beträgt. Die Analyse der Ladungsdichteverteilung und Berechnung des Ladungstransfers an den Grenzflächen ergab, dass ein ionischer Beitrag von den Nb-O-Bindungen der Grenzfläche die Bindung verstärkt. Zudem ist im Gegensatz zu den anderen Metallen bei bei Niob das d-Band nur teilweise gefüllt, was ebenfalls zu stärkeren Grenzflächenbindungen führt.
|
|
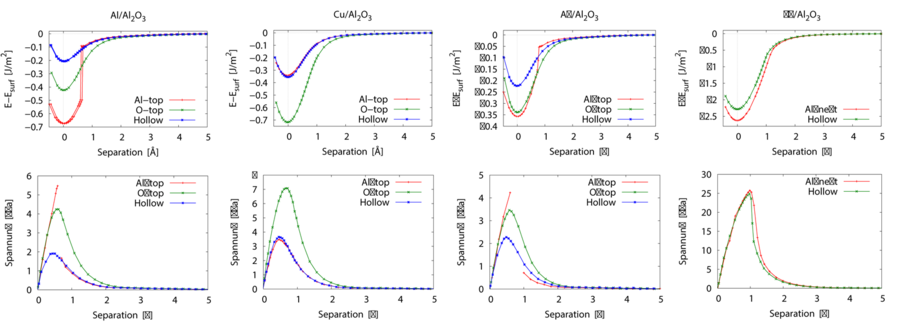
Da in der Realität aufgrund von Defekten oder mechanischer Belastung Gitterverzerrungen auftreten, wurde bei diesen Grenzflächensystemen auch untersucht, wie sich Separationsarbeit und Maximalspannung bei Dehnung oder Stauchung des Systems in Grenzflächenebene ändern. Bei Dehnung wurde eine Absenkung von Separationsarbeit und Maximalspannung gefunden während diese Werte bei Stauchung zunehmen. Besonders deutlich ist dieser Effekt bei den Metallen, deren grenzflächennächste Atomschicht nach der Relaxation nicht mehr planar ist.