
Ergebnisse
Proteindarstellungen
|
|
|
|
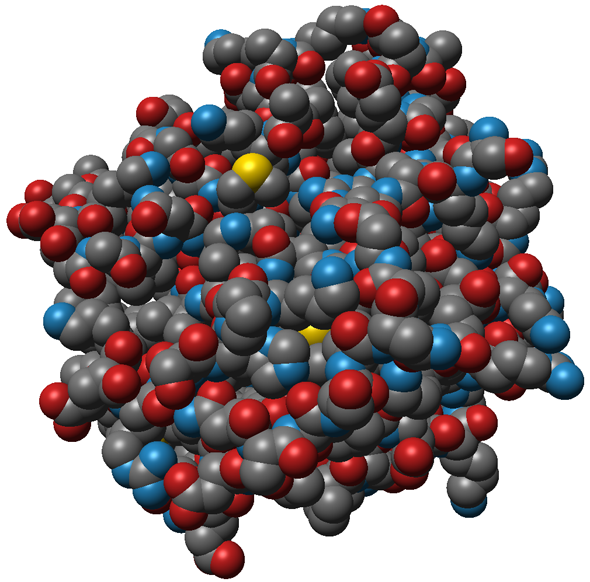
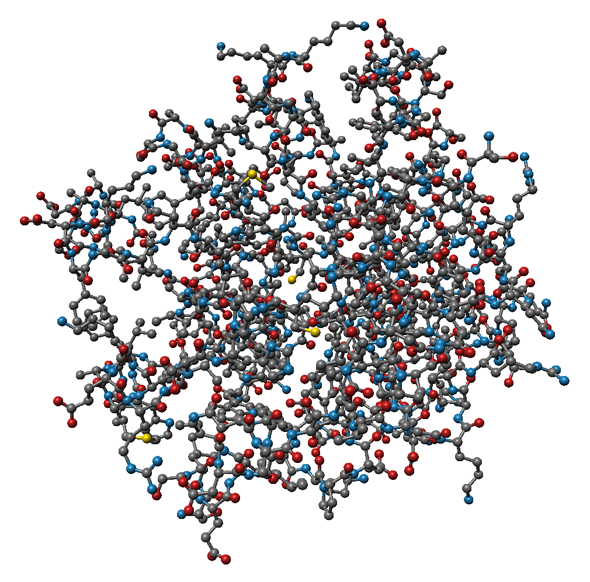
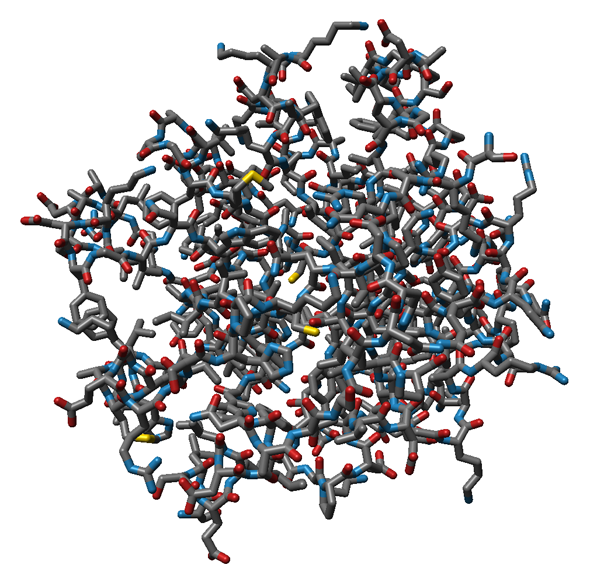
Die interaktive Visualisierung von Proteinen und anderen (Bio-)Molekülen ist ein wichtiges Werkzeug für die visuelle Analyse insbesondere zeitabhängiger Daten, welche das Ergebnis von Molekulardynamik- (MD-)Simulationen sind. Diese auch als Trajektorien bezeichneten Datensätze können mehrere zehn- bis hunderttausende von Zeitschritten einer Simulation enthalten und mehr als 20 GB Speicher benötigen. Zu diesem Zweck wurden herkömmliche Proteindarstellungen mit zeitgemäßer Optik implementiert, wobei die Möglichkeiten aktueller Hardware ausgenutzt wurden, um eine interaktive Darstellung zu ermöglichen. Bei den bisher unterstützten atombasierten Moleküldarstellungen (siehe Abbildung 1) werden punktbasierte Glyphen mittels GPU-Raycasting gerendert, um hohe Darstellungsqualität bei interaktiven Frameraten zu gewährleisten.
|
|

Die Cartoon- oder Ribbon-Darstellung ist eine abstrakte Visualisierung, welche die Sekundärstruktur eines Proteins zeigt (siehe Abb. 2). Diese entsteht durch Faltung der Aminosäureketten in die sogenannten Sekundärstrukturelemente Helix, Sheet, Turn und Random-Coil. Die in diesem Teilprojekt entwickelte Implementierung bietet die Möglichkeit, die Geometrie komplett im Geometry Shader der Graphikkarte zu erzeugen, um auch bei großen Trajektorien eine hohe Anzeigegeschwindigkeit zu gewährleisten. Details sind in "GPU-based Visualisation of Protein Secondary Structure" zu finden.
|
|
|
|
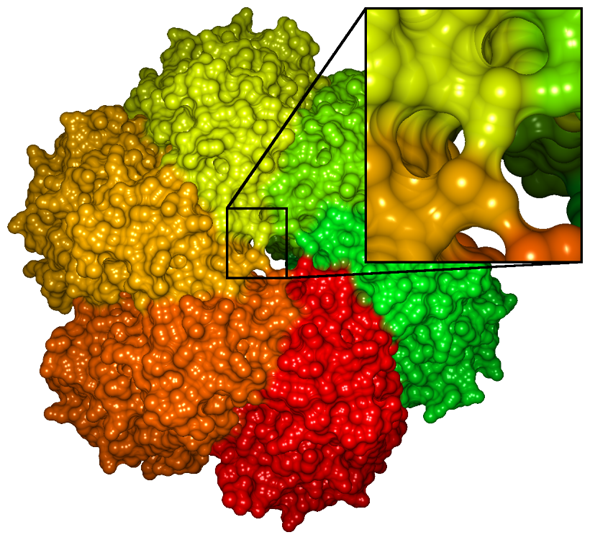
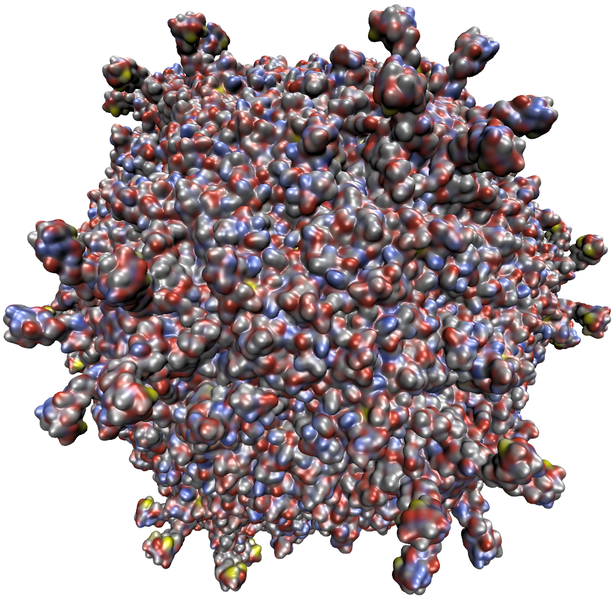
Oberflächendarstellungen von Proteinen werden benötigt, wenn die Kontaktfläche zwischen einem Protein und dem umgebenden Medium (Lösungsmittel) bzw. anderen Molekülen untersucht wird. Typische Anwendungen sind docking (die Analyse der Interaktion des Proteins mit anderen Molekülen) und die Untersuchung spezifischer Merkmale, welche an der Oberfläche auftreten (beispielsweise Bindungsstellen oder hydrophobe/-phile Regionen), aber auch die Untersuchung innerer Strukturen wie Hohlräumen im Protein. Die Solvent Excluded Surface (SES) ist die gebräuchlichste Oberflächendarstellung, da sie für alle oben genannten Anwendungen geeignet ist (siehe Abb. 3).
Durch die aufwändige Vorberechnung stellt die interaktive Visualisierung der SES insbesondere für große Trajektorien eine Herausforderung dar. Eine erste im Rahmen des SFB entwickelte Implementierung kombinierte eine abschnittsweise Aktualisierung der Oberfläche mit GPU-Raycasting punktbasierter Glyphen, um dieses Ziel zu erreichen. Des Weiteren wurde insbesondere für große Proteine eine semantische Reduktion der Atomdaten vorgeschlagen, welche für höhere Frameraten sorgt. Im weiteren Projektverlauf wurden Implementierungen entwickelt, welche die komplette Berechnung der SES auf die Graphikkarte auslagern, um eine die Berechnung zu beschleunigen. Die entwickelten Methoden werden in "Interactive Visualization of Molecular Surface Dynamics", "Parallel Computation and Interactive Visualization of Time-varying Solvent Excluded Surfaces" und "Parallel Contour-Buildup Algorithm for the Molecular Surface" erläutert.
Mit den oben genannten Methoden ist es möglich, die SES für Moleküle mit bis zu 10.000 Atomen interaktiv zu berechnen und darzustellen. Für größere Daten wurde deshalb gemeinsam mit Wissenschaftlern der University of Illinois at Urbana-Champaign eine geeignete Approximation der SES entwickelt, welche ebenfalls komplett auf der Graphikkarte berechnet wird (siehe Abb. 4). Mit dieser Darstellung ist es möglich, die Oberfläche von Molekülen mit mehr als einer Million Atomen interaktiv zu visualisieren (siehe "Fast Visualization of Gaussian Density Surfaces for Molecular Dynamics and Particle System Trajectories").
Lösungsmitteldarstellungen
|
|
|


Lösungsmittel haben einen wichtigen Einfluss auf die Funktion des Proteins. Darum ist es von großem Interesse, das Verhalten des Lösungsmittels zu analysieren.
Durch die Berechnung und Visualisierung von Lösungsmittelpfaden können neben den sogenannten Stellen kondensierten Wassers auch die prinzipiellen Pfade, welche die Lösungsmittelmoleküle durch Bindungstaschen nehmen, untersucht werden. Die Anwendung wird in "Visual Abstractions of Solvent Pathlines near Protein Cavities" beschrieben.
Ein wichtiges Werkzeug bei der Darstellung und Analyse des Lösungsmittels sind Filter, wodurch beispielsweise nur die Lösungsmittelmoleküle dargestellt werden, welche mit dem Protein oder einem anderen Polymer interagieren (siehe "Interactive Exploration of Polymer-Solvent Interactions"). Eine zeitliche Aggregation der Lösungsmittelmoleküle oder der Ionen im Lösungsmittel kann außerdem die Analyse der gemittelte Bewegungsrichtung oder -geschwindigkeit ermöglichen (siehe "Visual Analysis for Space-Time Aggregation of Biomolecular Simulations").
Analyse und Visualisierung von Cavities
|
|
|
|
|
|

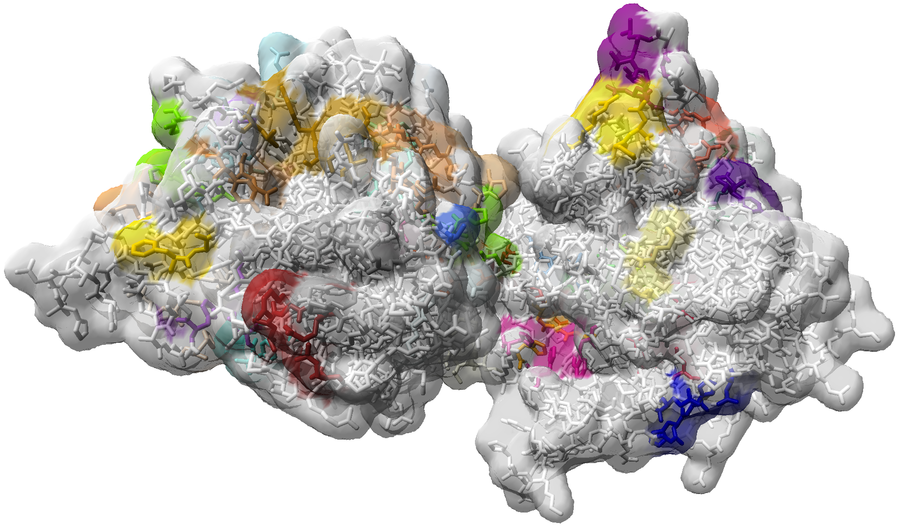
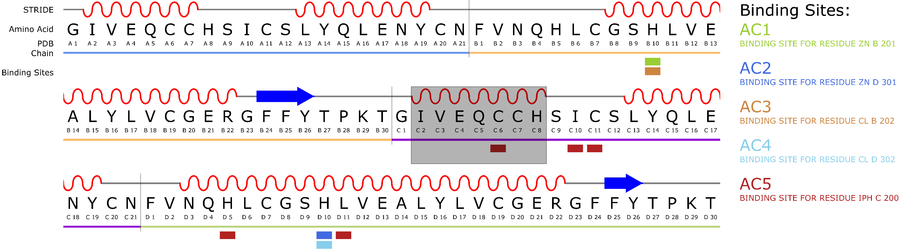
Die Struktur eines Proteins spielt eine wichtige Rolle für seine Funktion. Neben der oben erwähnten Sekundärstruktur ist auch die räumliche Struktur der Moleküloberfläche essentiell. Die Moleküloberfläche stellt das Interface zwischen einem Protein und umgebenden Molekülen dar. In mehreren, aufeinander aufbauenden Arbeiten wurden zunächst Hohlräume innerhalb des Proteins untersucht, dann Einbuchtungen der Oberfläche (sogenannte cavities) und schließlich die Zugänglichkeit von Bindungsstellen und Kanälen. Die cavities (Hohlräume, Bindungstaschen und Kanäle) werden dabei auch zeitlich nachverfolgt, um Veränderungen während der Simulation zu erkennen. Die gesamte Berechnung ist GPU-beschleunigt und läuft in Echtzeit während der Visualisierung. Somit ist eine explorative visuelle Analyse durch einen Experten möglich. Die Analyse wird zusätzlich durch Diagramme und Graphen unterstützt, welche die stationären und extrahierten, dynamischen Eigenschaften der simulierten Moleküle darstellen. Die entstandenen Methoden und Anwendungen werden in "Interactive Exploration of Protein Cavities", "Interactive Extraction and Tracking of Biomolecular Surfaces Features" und "Visual Analysis of Dynamic Protein Cavities and Binding Sites" beschrieben.
Visual Analytics von Protein-Ensembles
|
|
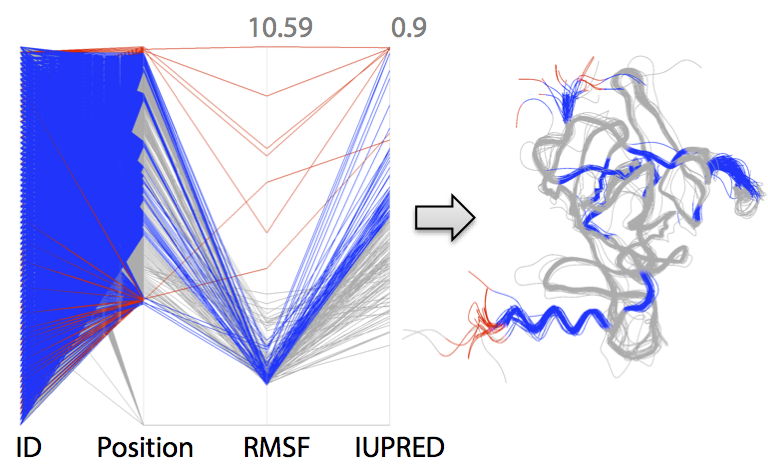
In Kooperation mit Teilprojekt D.5 und Wissenschaftlern des CSIRO in Australien wurde eine Visual-Analytics-Applikation zur Untersuchung von Protein-Ensembles umgesetzt. Ensembles vergleichbarer Proteine können zum Beispiel entweder aus einer Datenbank stammen oder die Ergebnisse verschiedener Simulationen sein. Ziel der Anwendung ist es, dem Benutzer viele zusätzliche Informationen zu den einzelnen Proteinen des Ensembles zu geben, welche durch verschiedene Analysemethoden extrahiert werden. Mit Hilfe einer Darstellung als Parallele Koordinaten soll der Benutzer korrelierte oder antikorrelierte Werte erkennen können und uninteressante Wertebereiche filtern. Das Resultat wird gleichzeitig in einer überlagerten 3D-Darstellung des Protein-Ensembles visualisiert (siehe Abb. 11). Weitere Informationen finden sich in "Visualising Intrinsic Disorder and Conformational Variation in Protein Ensembles". Eine ausführbare Testversion für Windows-Betriebssysteme kann hier heruntergeladen werden.
MegaMol-Demonstrator
Alle im Rahmen von Teilprojekt D.4 entstandenen Visualisierungen wurden als MegaMol-Plugin implementiert. MegaMol ist ein gemeinsam genutzes Visualisierungs-Framework, welches im SFB 716 von Teilprojekt D.3 entwickelt wurde. Mehr Informationen und eine vorkonfigurierte Demo-Version einiger Protein-Visualisierungen finden sich auf der MegaMol-Entwicklerseite unter www.visus.uni-stuttgart.de/megamol/wiki/Protein.
Kartenbasierte Visualisierung von Proteinoberflächen
Bei der Analyse bereits verfügbarer Abbildungen molekularer Oberflächen (vgl. Abb. 3,4) fällt eine Problematik der Repräsentation auf: Sowohl die verwundenen Strukturen der Oberfläche, als auch deren Dreidimensionalität verhindern eine sofortige Wahrnehmung der kompletten Oberfläche. Daher wurde eine Methode zur Generierung zweidimensionaler Oberflächenrepresentationen entwickelt. Dazu wird zunächst die bereits bekannte Proteinoberfläche in eine Kugel deformiert, welche dann benutzt wird, um eine Karte der Oberfläche zu generieren.
 |
|
Dies geschieht unter Zuhilfenahme bekannter Projektionsmethoden aus der Karthografie, wie zum Beispiel der Mercator-Projection. Weitere Details können "Molecular Surface Maps" entnommen werden.
 |
|
Unsicherheitsvisualisierung für Protein-Sekundärstrukturen
Die Cartoon-Darstellung (vgl. Abb. 2) ist eine populäre Visualisierungsmethode für Proteine, da sie sowohl die Sekundär-strukturen als auch die räumliche Anordnung darstellt. Typischerweise kann zusätzlich durch die Benutzung von Farbe noch ein weiterer Wert dargestellt werden. Wenn aber ein zweiter Wert, wie zum Beispiel Unsicherheit, hinzugenommen wird, gelangen bereits existierende Visualisierungsmethoden an ihre Grenzen. Daher wurde ein Verfahren entwickelt, um weitere Werte in die bereits bekannte Cartoon-Darstellung einbringen zu können.
Dies geschieht durch Modifikation der Oberflächenwelligkeit. Basierend auf der Größe des dargestellten Wertes wird sowohl die Frequenz als auch die Höhe der Oberfläche modifiziert. Die resultierende Visualisierung erlaubt nun einen einfachen Vergleich der neu hinzugefügten Größen. Die Arbeit "Uncertainty Visualization for Secondary Structures of Proteins" führt weitere Details hierzu aus.