
Results
Development of massively-parallel MD software
In cooperation with the subproject D.1, the molecular dynamics (MD) code ls1 Moldy was further optimized and expanded with respect to new functionalities. The necessary changes were so significant that an entirely new concept had to be chosen and implemented. This work was done in cooperation with the chair SCCS at TU München (Prof. H.-J. Bungartz). However, the framework remained the same: MD that was optimized to large numbers of particles on massively-parallel computing hardware. The result of this work is new software ls1 Mardyn that considers interaction sites of Lennard-Jones (LJ) and point polarity type. This approach is well suited for very accurate description of pure fluids and mixtures thereof. Furthermore, the Tersoff-Potential was implemented as well to consider solid nanostructures in contact with the fluid that allows studying their influence on the fluid properties. On the basis of ls1 Mardyn, both equilibrium and non-equilibrium states can be simulated. For Couette and Poiseuille flows, controlled external force was implemented that allows specifying an average flow velocity.
|
|
|
|
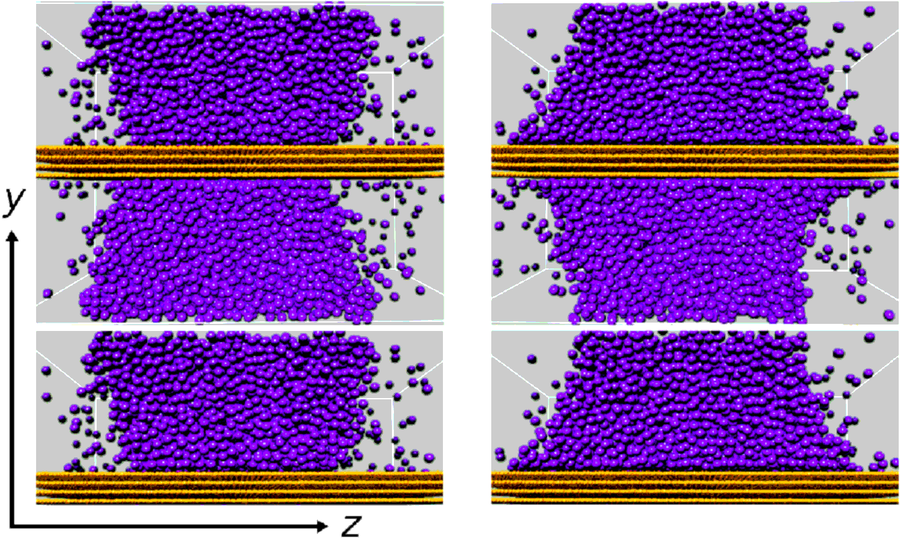
Multi-phase fluid in nanochannels
The main topic of this project was to quantify the influence of the interaction between the solid wall atoms on the fluid molecules on the structural and dynamic properties of fluids near walls. In this context, the contact angle is of particular interest, because it can be well accessed experimentally and because it characterizes the influence of a wall on the fluid. Within the project, the influence of the unlike dispersive interaction between wall and fluid was studied for the truncated and shifted LJ fluid. If that potential is assumed between the fluid molecules as well as fluid molecules and wall atoms, then there is a value for the reduced unlike dispersive interaction energy for which the contact angle is 90°, independent on the temperature. The dependence of the contact angle on the unlike dispersive energy is symmetric with respect to this value. Among others, these results may well be used to estimate appropriate potential parameters for given fluid-wall material pairs on the basis of contact angles. These investigations were made in close cooperation with project A.2, because they also allow for parameter mapping between MD and lattice Boltzmann simulations.
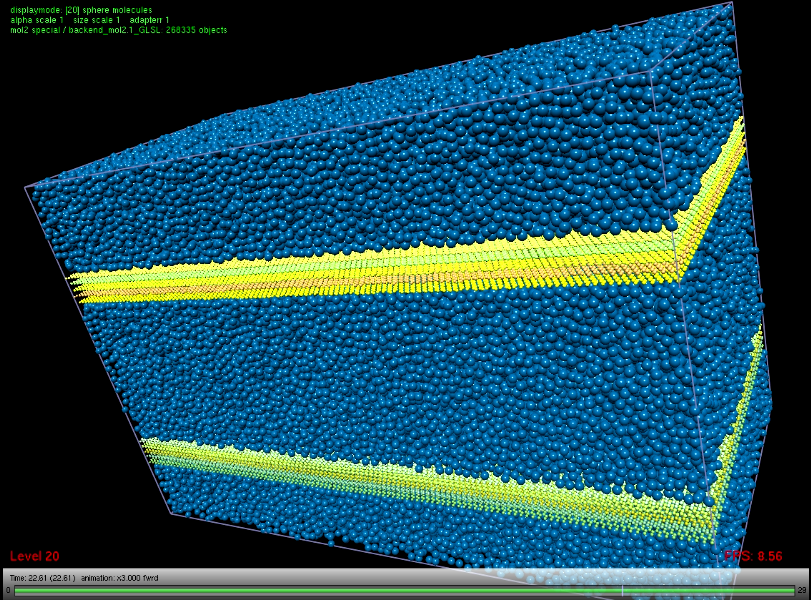
MS simulation of nanoscale flows
Couette and Poiseuille flows, induced by a controlled external force, were studied in certain cases for cylindrical and more complex wall geometries. Systematic studies were made for planar slit geometries. Carbon nanotubes and graphite were considered as wall materials, methane was chosen as fluid. The goal was to determine the dependence of the pressure loss and the wall slip properties on the channel diameter. On the basis of the fluid velocity profile, aggregated properties like the slip length can be calculated and used e.g. as boundary conditions for continuum solvers. The pressure loss was obtained directly via the force that was applied to maintain the flow. For the considered systems, the critical diameter was found to start around 50 nm, smaller systems are dominated by the wall slip, larger systems exhibit less influence by the wall. The range for which the wall slip is paramount for this pair of material was entirely covered by the present MD simulations, the range where the flow profile converges with the classical solution was partly covered. The transition through that critical range does not lead to a specific behavior of the pressure loss, which was found to be inversely proportional to the cross-section area (Darcy’s law).
Equilibrium properties of curved interfaces
For non-planar nanoscale interfaces, the curvature of interfaces is so pronounced that all its properties are significantly influenced. The thickness of the interface is non-negligible in comparison to the curvature radius so that a description by an interface surface is not appropriate anymore. In this case also the third spatial direction orthogonal to the interface has to be considered, because many thermophysical properties can be analyzed only along the interface profile. This also holds to some extent for the surface tension. Theoretical results and molecular simulations show that the surface tension approaches zero for a vanishing curvature radius. The vapor pressure of the droplet is influenced by several effects that partly cancel each other out, thus it is in good agreement with the classical nucleation theory that does not consider any of these effects. In comparison to a droplet that consists of an incompressible liquid, the reduced surface tension is associated with an increase surface of tension.
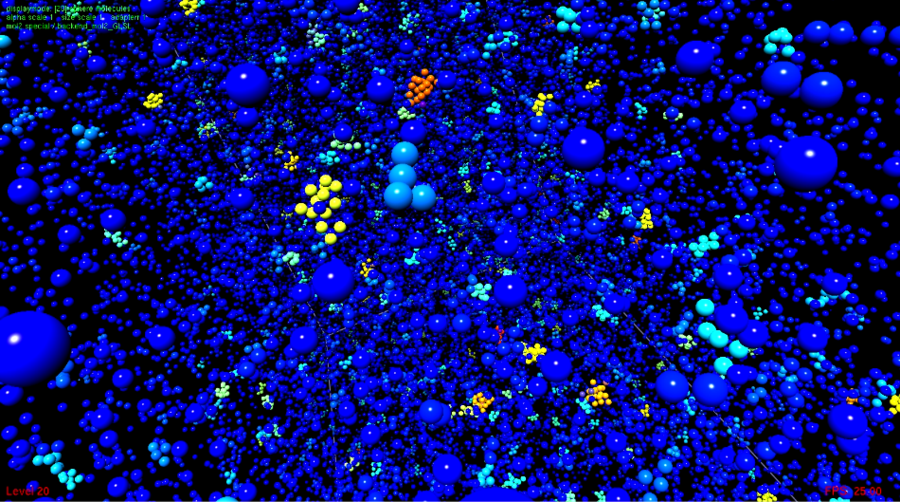
Homogeneous nucleation in supersaturated vapors
The homogeneous nucleation in supersaturated vapors that is the main application of the classical nucleation theory was studied by MD simulation. Together with the subprojects D.1 and D.3, a comparison of cluster criteria that define emerging droplets of the molecular scale was made with respect to their applicability in MD. In cooperation with project D.3, appropriate visualization methods were developed, the institute VIS implemented them into the MolCloud code. The central aim was to intuitively understand the aspects of the nucleation process and also to support the assessment of different cluster criteria. Nucleation is a non-equilibrium process that leads to the breakdown of a metastable state. To study this process, a new method was proposed that combines grand canonical MD simulation with a variant of Maxwell’s demon that was theoretically discussed by McDonald: Interventions of this demon extract the largest droplets out of the system and conserve thereby its state. At the same time, this approach allows for the direct determination of the nucleation rate, i.e. the number of emerging droplets per volume and time, which is the characteristic property of this process. In comparison to experimental work, where only relatively low supersaturations can be studied, the present work showed a good agreement with the classical nucleation theory for highly supersaturated states. This holds both for the truncated and shifted LJ fluid and the quadrupolar fluids ethane and carbon dioxide.